
Die Krankheit der 1000 Gesichter
- katrinflatscher
- 28. Feb. 2025
- 11 Min. Lesezeit

Wenn man Kenan zum ersten Mal trifft, dauert es maximal 10 Minuten, bis man völlig vernarrt in ihn ist. Das mag an seinem strohblonden Haarschopf liegen, der einen sofort an Michel von Lönneberga aus Astrid Lindgrens beliebter Kinderbuchreihe denken lässt. Oder an seinen leicht abstehenden Ohren, die ihn ein bisschen wie einen schelmischen Kobold wirken lassen, der gerade einen Streich ausheckt. Vielleicht ist es auch sein spitzbübisches Lächeln mit den paar großen Zahnlücken, wo die Milchzähne bereits ausgefallen sind, wie das in dem Alter eben typisch ist. Ebenfalls typisch ist Kenans Begeisterung für Superhelden, Drachen und Ritter. Stolz zeigt er mir sein Halskettchen, an dem an einem dünnen Lederband ein kleiner Schwert-Anhänger baumelt. Soll heißen: „Schau her, auch ich bin ein starker Kämpfer, genauso wie ein echter Superheld!“
Es ist gut, dass Kenan ein starker Kämpfer ist. Mit seinen knapp 7 Jahren weiß er es noch nicht, aber innere Stärke ist eine Eigenschaft, die er in seinem Leben wohl gut gebrauchen wird können. Was man nämlich nicht auf den ersten Blick sieht, wenn man den quirligen kleinen Jungen beim Spielen beobachtet, ist dass er an einer unheilbaren seltenen Krankheit leidet.
Wie der Name schon vermuten lässt, sind seltene Krankheiten ausgesprochen rar. Sie betreffen nur einen Bruchteil der Bevölkerung - in der EU definitionsgemäß weniger als eine von 2000 Personen. Das bedeutet aber nicht, dass sie nicht weit verbreitet wären, denn insgesamt gibt es rund 6.000 verschiedene seltene Krankheiten, die in der Medizin derzeit bekannt sind. Alleine in Europa sind rund 30 Millionen Menschen, also ungefähr 4-6% der Gesamtbevölkerung, von irgendeiner davon betroffen. Da gibt es zum Beispiel das Marfan-Syndrom, eine Bindegewebsstörung, die das Herz, die Augen und das Skelett betrifft, die tückische Progerie, bei der Betroffene bereits als Kleinkinder übermäßig schnell altern, oder das Stiff-Person-Syndrom, das in den letzten Jahren zumindest etwas bekannter geworden ist, seit die kanadische Sängerin Celine Dion öffentlich gemacht hat, dass sie daran leidet.
Kenans Krankheit findet man auf der langen Liste der seltenen Krankheiten unter dem Buchstaben N. N wie Neurofibromatose, manchmal auch von-Recklinghausen-Krankheit genannt. Als seine Eltern diesen Begriff zum ersten Mal lesen, ist Kenan noch nicht mal ein halbes Jahr alt. Seiner Mutter Miriam fällt in seinen ersten Lebensmonaten auf, dass Kenan einige Pigmentflecken auf Bauch und Rücken hat, die sich farblich deutlich von der zartrosa Babyhaut abheben. Da sie sich nicht erklären kann, woher sie kommen, recherchiert sie arglos, was es damit auf sich haben könnte. Im Internet stößt sie auf die Bezeichnung „Café-au-lait-Flecken“ – eine genau passende Beschreibung für die milchkaffeefarbenen Pigmentierungen auf Kenans Körper. Die Flecken seien aber keine Muttermale oder irgendeine Form von Hautausschlag, liest sie, sondern könnten ein frühes Anzeichen von Neurofibromatose sein – einer genetischen Krankheit, von der sie noch nie zuvor gehört hat. Auch ihr Mann Christian, zufällig selbst von Beruf Genetiker, kann mit dem Begriff nur wenig anfangen. Die Informationen und vor allem die Bilder, die das Internet dazu ausspuckt, sind aber mehr als beunruhigend, und so beschließen sie, die Sache beim nächsten Arzttermin abklären zu lassen.
Der Kinderarzt, bei dem sie ihren Verdacht äußern, wiegelt ab. Auch er als Mediziner hat noch nie mit einem Neurofibromatose-Patienten zu tun gehabt. Dass Hautflecken ein Indiz für eine genetische Krankheit sein könnten, kann er sich nicht vorstellen. Zur Sicherheit verweist er die Familie aber an einen Neuropädiater. Dieser bestätigt, dass es tatsächlich einen bewiesenen Zusammenhang zwischen einer gewissen Fleckenzahl und der Krankheit Neurofibromatose gibt. Für eine gesicherte Diagnose brauche es allerdings noch zusätzliche Symptome oder ein betroffenes Elternteil, von dem die Krankheit vererbt worden sein könnte.
Die Begriffe „genetische Krankheit“ und „Erbkrankheit“ werden oft synonym verwendet. Tatsächlich gibt es zwischen ihnen aber einen Unterschied: Alle Erbkrankheiten sind genetisch, aber nicht alle genetischen Krankheiten sind vererbbar. Genetische Krankheiten können auch spontan entstehen, z. B. durch zufällige Mutationen bei der Zellteilung oder durch schädliche Umwelteinflüsse. Die resultierenden Veränderungen in der DNA werden aber normalerweise nicht an die Nachkommen weitergegeben.
Mutationen, die bereits in den Keimzellen (den Eizellen oder Spermien) vorhanden sind, werden dagegen häufig vom betroffenen Elternteil an das Kind vererbt. Die damit in Verbindung stehenden Krankheitsanzeichen manifestieren sich dann bereits von Geburt an oder im frühen Kindesalter. Etwa die Hälfte der Neurofibromatose-Fälle entsteht auf diese Weise. Das mutierte Gen wird autosomal-dominant vererbt: Das bedeutet, dass ein Kind eine 50-prozentige Chance hat, ebenfalls Neurofibromatose zu bekommen, wenn bereits Vater oder Mutter die Erkrankung haben. In der anderen Hälfte der Fälle passiert die Mutation allerdings völlig zufällig im Zuge der Embryonalentwicklung. Bekannte Risikofaktoren gibt es nicht.
In Kenans Familie ist niemand von Neurofibromatose betroffen. Vererbt worden kann die Krankheit also nicht sein. Um dennoch Gewissheit zu bekommen, entscheiden sich seine Eltern für einen Gentest. Nach mehreren Wochen bangen Wartens bestätigt sich der schlimme Verdacht: Kenan hat tatsächlich die Mutation im betreffenden Gen. Diagnose: Neurofibromatose Typ 1, kurz: NF1.
Die Krankheit kann sich aber nicht nur durch Pigmentstörungen äußern. Tatsächlich sind die Café-au-lait-Flecken das einzige Symptom von NF1, das völlig harmlos ist. Wesentlich gefährlicher ist das, was im Körper der Betroffenen passiert.

Kenan leidet an Neurofibromatose Typ 1 (Privataufnahme)
Bei Neurofibromatose wird ein wichtiges Tumorsuppressorprotein namens Neurofibromin verändert oder ganz ausgeschaltet. Normalerweise haben Tumorsuppressoren die Aufgabe, das normale Zellwachstum im Körper zu regulieren und die Entstehung von Tumoren zu unterdrücken. Ist aber das Gen, das die Neurofibromin-Produktion steuert, durch eine Mutation gestört, funktioniert diese Regulation nicht mehr richtig. Zellen und Gewebe können dann unkontrolliert wachsen – auf und unter der Haut, an den Augen, an den Knochen und an allen anderen Organen, durch die Nervenfasern verlaufen.
Ob, wo und wann diese Tumore auftreten, kann man nicht vorhersagen, sondern lediglich, dass eine hohe Neigung dazu besteht. Deswegen nennt man Neurofibromatose im medizinischen Fachjargon auch ein Tumorprädispositionssyndrom. Für Betroffene bedeutet das, dass sie ständig auf der Hut und aufmerksam für Veränderungen im Körper sein müssen. Manche haben zeitlebens nur milde Symptome, die sie kaum im Alltag beeinflussen. Andere erleben einen schweren Verlauf mit starken Komplikationen und massiven Einschränkungen. Aufgrund der unzähligen unterschiedlichen Ausprägungen wird Neurofibromatose auch manchmal „die Krankheit der tausend Gesichter“ genannt.
Der selbst von NF1 betroffene kolumbianische Arzt Juan Sebastián Botero-Meneses beschreibt die Krankheit in einem Fachbeitrag im Permanente Journal als sprichwörtliches Damokles-Schwert. In Ciceros Anekdote ist Damokles ein Günstling des Tyrannen Dionysios von Syrakus, der neidisch auf dessen Reichtum und Macht ist. Um ihm eine Lektion zu erteilen, bietet der Herrscher Damokles an, mit ihm die Plätze zu tauschen. Er lässt ihn auf seinem Thron sitzen, über dem ein großes Schwert an einem einzelnen Rosshaar aufgehängt worden ist. Das Schwert kann jeden Moment auf Damokles herabfallen und ihn durchbohren – oder auch nicht. Das Bild ist heute eine Parabel für eine ständig drohende Gefahr, bei der man nie genau weiß, ob und wann sie eintritt.
„Wenn er im Krankenhaus ist, dann hab ich immer Sorgen, dass was in ihm drin ist.“, beschreibt es Jasmin, Kenans ältere Schwester, treffend. Sie kennt die Abläufe rund um die regelmäßigen Vorsorgeuntersuchungen ihres Bruders mittlerweile bestens und weiß, dass alle Familienmitglieder dann stets bangen, dass hoffentlich keine Auffälligkeiten gefunden werden. Mindestens zweimal jährlich muss sich Kenan diesem Untersuchungsmarathon unterziehen. Die zahlreichen Tests nehmen mehrere Tage in Anspruch, die er zusammen mit seiner Mutter stationär im Krankenhaus verbringt. Am wichtigsten ist dabei die Magnetresonanztomografie (kurz: MRT), mit der man Feinstrukturen im Körper präzise sichtbar machen und entstehende Tumore besser erkennen kann als mit anderen Techniken. Die Untersuchung ist zwar völlig schmerzfrei, aber ziemlich unangenehm. In einer engen Röhre muss man mehrere Minuten perfekt stillliegen und den hohen Geräuschpegel des Geräts über sich ergehen lassen. Schon für Erwachsene ist das oft eine große Herausforderung, für kleine Kinder ist es kaum schaffbar, weshalb die Untersuchung meist nur unter Narkose durchgeführt werden kann.
Die überwiegende Mehrzahl der Tumore bei NF1 ist gutartig. Das heißt aber nicht, dass sie harmlos sind. Da sie auf nahezu allen Nerven im Körper wachsen können, ist es nicht selten, dass sie auf umliegendes Gewebe drücken oder benachbarte Organe beeinträchtigen können. Besonders kritisch ist das am feinstrukturierten Sehnerv, der tief in der Schädelhöhle eingebettet verläuft und in unmittelbarer Nähe zum Gehirn liegt. Entsteht dort ein Tumor, ein sogenanntes Optikusgliom, kommt man ihn so gut wie nicht an ihn heran, um ihn chirurgisch zu entfernen. Stattdessen muss man dann versuchen, das Gliom mittels Chemotherapie schrumpfen zu lassen oder zumindest am Weiterwachsen zu hindern.
Zu den augenärztlichen Untersuchungen kommen noch viele weitere Tests, denen sich NF1-Patienten regelmäßig unterziehen müssen. Orthopäden untersuchen die Knochenstruktur, denn zahlreiche Betroffene entwickeln Verkrümmungen der Wirbelsäule, Verformungen der Knochen oder „Falschgelenke“. Neurologen prüfen Reflexe, Muskelkraft und Sensibilität und beurteilen die kognitive und psychologische Entwicklung der Patienten. Dermatologen begutachten Hautauffälligkeiten, um möglichst frühzeitig festzustellen, ob sich irgendwo Gewebeveränderungen bilden, die behandelt werden müssen.
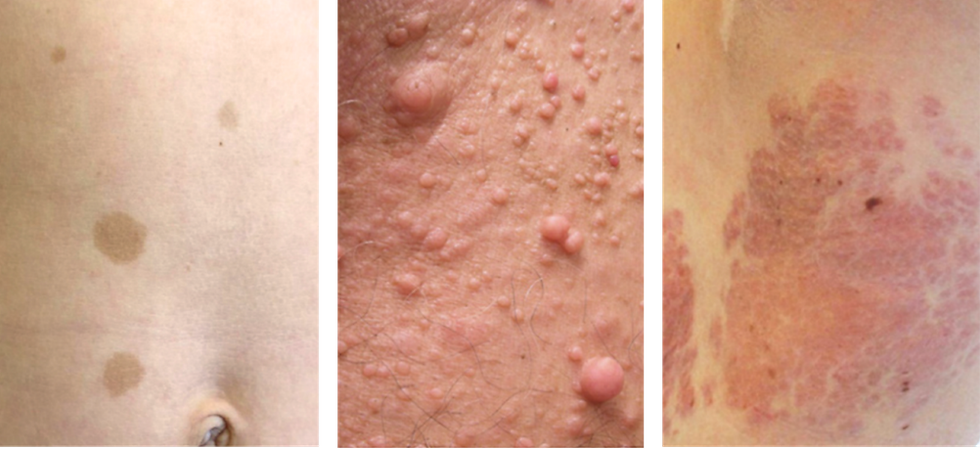
Neurofibromatose-Symptome auf der Haut (Café-au-lait-Flecken, Neurofibrome, plexiformes Neurofibrom), Bild © A. Azizi
Die namensgebenden Neurofibrome bilden sich bei fast allen Betroffenen über kurz oder lang. Dass sie auf oder direkt unter der Haut wachsen, ist in diesem Fall ein großer Vorteil, denn, so zynisch das auch klingen mag: nach außen ist wenigstens Platz. Für die Patienten sind sie allerdings einer der belastendsten Krankheitsaspekte, denn oft sind sie deutlich sichtbar – etwa auf den Armen, am Hals oder im Gesicht. Viele Betroffene fühlen sich dadurch extrem unsicher und meiden soziale Situationen, weil sie fürchten, unangenehmen Reaktionen ausgesetzt, angestarrt oder womöglich sogar beleidigt zu werden. Fibrome können außerdem einen hohen Leidensdruck verursachen, wenn sie jucken, schmerzen oder an empfindlichen Körperstellen sitzen. Manchmal können sie mit kleinen Eingriffen oder mittels Laser entfernt werden, doch die Rückkehrquote ist hoch.
Manche Menschen entwickeln nur wenige Fibrome, andere Dutzende oder sogar Hunderte davon. Besonders gefürchtet sind plexiforme Neurofibrome, eine besondere Form von Tumoren, die flächig wachsen und oft große, weiche Wucherungen bilden. Sie können in der Haut, aber auch um Organe, Nerven oder Knochen herum wachsen und je nach Anzahl und Lage überall im Spektrum von „kaum auffallend“ bis zu „schwer entstellend“ liegen.
Der Protagonist des preisgekrönten Films „A different man“, Adam Pearson, veranschaulicht das sehr deutlich. Der Schauspieler, der im Film die Rolle des Oswald spielt, leidet selbst an Neurofibromatose und hat eine besonders schwere Ausprägung entwickelt, durch die sich mit der Zeit sein Gesicht stark verformt hat. Viele seiner Fibrome sind mit wichtigen Nerven und Gefäßen verwachsen, sodass sie nicht gefahrlos entfernt werden können. Nach zahlreichen Operationen hat Pearson sich bewusst dazu entschieden, auf weitere Eingriffe zu verzichten, um nicht sein verbleibendes Sehvermögen zu riskieren.
Bezeichnend an seinem Fall ist auch, dass sein eineiiger Zwillingsbruder Neil ebenfalls von der Krankheit betroffen ist, jedoch im Vergleich zu Adam so gut wie keine Fibrome entwickelt hat. Die Brüder sind nur zwei der Tausenden Gesichter, die Neurofibromatose haben kann. Beide setzen sich heute aktiv für Aufklärung über Neurofibromatose und für den Umgang mit sichtbaren Behinderungen in der Gesellschaft ein.

Die Brüder Neil & Adam Pearson (Bild James Newton Films, ,CC BY-SA 3.0 via Wikimedia Commons)
Menschen mit seltenen Krankheiten stehen im Alltag immer wieder vor Herausforderungen, die über die medizinischen Symptome hinausgehen. Das liegt vor allem am mangelnden Bewusstsein in der Gesellschaft. Bekannte, Freunde, Lehrer oder Arbeitgeber, ja selbst viele Ärzte, können sich unter der jeweiligen Krankheitsbezeichnung nichts vorstellen und können nicht einschätzen, was die Diagnosen im Alltag bedeuten.
Im sozialen Umfeld müssen Betroffene immer wieder aufs Neue erklären, was ihre Erkrankung eigentlich ist, wie sie sich äußert und - vor allem wenn sie wie Neurofibromatose mit auffälligen äußeren Symptomen einhergeht - dass sie nicht ansteckend ist. Das kann emotional extrem anstrengend sein, besonders wenn sie dabei auf Unverständnis, Vorurteile oder Ablehnung stoßen.
Gleichzeitig ist es eines der dringendsten Anliegen von Betroffenen, dass ihre seltenen Krankheiten mehr Aufmerksamkeit bekommen. Damit sind aber nicht neugierige Blicke, Sonderbehandlungen und Mitleidsbekundungen gemeint, sondern vor allem mehr Aufmerksamkeit seitens der Forschung. Nahezu alle seltenen Krankheiten haben das Problem, dass sie in der Grundlagenforschung kaum bedacht werden. Fördermittel für wissenschaftliche Projekte sind nur schwer zu bekommen, aussagekräftige Zell- und Tiermodelle fehlen und die Entwicklung gezielter Therapieansätze hinkt dem Bedarf meilenweit hinterher. In den meisten Fällen können nur die Symptome der seltenen Krankheiten behandelt werden, nicht aber ihre Ursachen. Die wenigen verfügbaren Medikamente sind oft extrem teuer. Eine einzelne Dosis des Medikaments Zolgensma, eines Gentherapeutikums gegen die seltene spinale Muskelatrophie, kostet beispielsweise über 2 Millionen US-Dollar.
Für Pharmaunternehmen ist es kaum rentabel, Medikamente für seltene Krankheiten, sogenannte Orphan Drugs, zu entwickeln. Um einen neuen Wirkstoff von der Forschungsphase bis zur Marktreife zu entwickeln, dauert es durchschnittlich 10-15 Jahre, und die Gesamtkosten belaufen sich auf ungefähr 2 Milliarden Euro - für nur ein einziges Medikament. Bei Blockbuster Drugs mit einem breiten Anwenderkreis, zum Beispiel bei Medikamenten gegen Bluthochdruck, sind diese enormen Entwicklungskosten wirtschaftlich gerechtfertigt, bei Medikamenten für einen winzigen Anwenderkreis aber nicht.
Dazu kommt, dass Orphan Drugs nicht auf reguläre Weise zugelassen werden können. Um die Wirksamkeit und Sicherheit neuer Medikamente zu prüfen, müssen diese normalerweise in mehrphasigen klinischen Studien an freiwilligen Probanden getestet werden. Diese müssen sich aber erst in ausreichender Zahl finden und zur Teilnahme bereit erklären. Gibt es weltweit nur ein paar wenige Betroffene, wie es bei seltenen Krankheiten der Fall ist, finden sich kaum genügend Testpersonen, um die für die Zulassung erforderliche statistische Sicherheit zu erreichen. Sind die Betroffenen zusätzlich Kinder, wird die Situation noch schwieriger.
Im Grunde wünschen sich Betroffene aber nicht nur wirkungsvolle Medikamente, um ihre Symptome in Schach zu halten, sondern realistische Möglichkeiten, ihre Krankheiten irgendwann vollständig zu besiegen und zu heilen. Die größte Hoffnung ruht auf modernen Zell- und Gentherapien wie der CRISPR/Cas9-Technologie, im Volksmund besser bekannt als die „Genschere“. Damit könnten die krankhaften genetischen Veränderungen korrigiert und durch fehlerfreie DNA-Abschnitte ersetzt werden - was eine faktische Heilung bedeuten würde. Noch stecken diese Technologien allerdings in den Kinderschuhen und sind bislang nur für wenige genetische Krankheiten klinisch erprobt. Sollten diese Therapien allerdings die erhofften Fortschritte machen, könnten sie den Durchbruch für die Heilung von Neurofibromatose und anderen selten Krankheiten bedeuten.

Im vergangenen Jahr ist Kenan eingeschult worden. Er besucht eine Integrationsklasse in der örtlichen Volksschule, wo geschulte Betreuungspersonen für Kinder mit besonderem Förderbedarf zur Verfügung stehen. Mit Neurofibromatose haben die Pädagogen dort aber auch noch nie zu tun gehabt. Auch im Bildungsbereich stehen seltene Krankheiten nicht auf der regulären Tagesordnung und es gibt keinen Standardplan für den Umgang mit den individuellen Bedürfnissen von betroffenen Kindern.
Kenan braucht vor allem Unterstützung bei seinen Aufmerksamkeitsschwierigkeiten und seiner Hyperaktivität - beides häufige Nebenerscheinungen von Neurofibromatose, die ins charakteristische ADHS-Spektrum fallen. Ruhig und konzentriert bei einer Sache zu bleiben fällt ihm schwer. Er möchte lieber herumrennen, klettern und seine Energie rauslassen als Buchstaben und das Einmaleins zu lernen. Bei Gesprächen plappert er oft dazwischen, meist mit der liebenswürdigen Eigenheit, die Rs als Ls auszusprechen und ungeduldig die Augen zu verdrehen, wenn man nicht sofort versteht, was er sagen möchte. Nicht selten ist er auch ein bisschen überschwänglich und unsanft, wenn er körperliche Nähe sucht oder mit anderen Kindern spielt. Neben der Schule bekommt er daher Unterstützung durch Ergotherapeuten, Logopäden und Kinderpsychologen, die ihn gezielt fördern und ihm dabei helfen, Entwicklungsverzögerungen auszugleichen. Dank der vielseitigen Hilfestellungen kann er im Moment noch ein kleiner Junge wie jeder andere auch sein.
Seine Krankheit ist Kenan noch nicht richtig bewusst. Doch er weiß, dass es bei den Gesprächen der Leute um ihn herum oft um ihn geht, um irgendwas mit Ärzten und Untersuchungsterminen. Als ich ihn frage, ob er Lust hat, ein Foto von sich zu machen, das er dann im Internet anschauen kann, zieht er instinktiv das T-Shirt hoch, um mir seine Café-au-lait-Flecken auf dem Bauch zu zeigen - in der Annahme, dass es sicher das sein muss, was mich als Fotomotiv interessiert. Am Ende machen wir dann ein paar Schnappschüsse davon, wie er aufgekratzt im Garten herumturnt. Er will natürlich alle Bilder sofort anschauen, um zu überprüfen, ob er darauf wirklich aussieht wie Spiderman oder ein anderer Superheld.
„Wenn man durch eine seltene Krankheit eines lernt, dann ist das, jeden Moment zu schätzen.“, sagt sein Vater Christian. „Im Moment geht es Kenan gut und es gibt keine kritischen Symptome, die unmittelbar behandelt werden müssen. Wir als Eltern können nur hoffen, dass das möglichst lange so bleibt. Und dass er immer die Fähigkeit behält, seine Aufmerksamkeit auf die kleinen, schönen Dinge im Leben zu lenken.“
Menschen mit seltenen Krankheiten entwickeln oft eine außerordentliche mentale Stärke und einen bemerkenswert resilienten Charakter. Vielleicht liegt das an den vielen Herausforderungen, die sie im Laufe ihres Lebens zu meistern lernen. Vielleicht stecken diese Eigenschaften aber auch von Anfang an in ihnen drinnen – wie eine besondere Mutation, die ihren Trägern die Fähigkeit verleiht, ihr Leben auf eine Weise zu leben, die den meisten anderen Menschen nur selten gelingt.
Als ich mich auf den Heimweg mache, ruft mir Kenan ein fröhliches „Tschüss Katlin!“ hinterher und schenkt mir zum Abschied noch ein besonders gewinnendes Zahnlückenlächeln. Für mich ist er jetzt schon ein kleiner Superheld und eine Art von Mensch, wie man sie quer durch alle Altersschichten nur selten trifft. Es stimmt wohl tatsächlich, was man sagt: Die seltensten Dinge auf der Welt sind auch die wertvollsten – nicht trotz, sondern gerade wegen ihrer Einzigartigkeit.
Quellen:
Botero-Meneses JS. The Sword of Damocles: Living with Neurofibromatosis Type 1. Perm J. 2020;24:19.111. doi:10.7812/TPP/19.111
Azizi A., Baumgartner A.-C. Neurofibromatose Typ 1 (NF1): Ein defektes Gen, viele verschiedene Gesichter. DFP Sonderpublikation. 2024. Springer Medizin
The Lancet Neurology. Rare diseases: maintaining momentum. Editorial. Lancet Neurol. 2022;21(3):203. doi:10.1016/S1474-4422(22)00046-1
Roessler HI, et al. Drug Repurposing for Rare Diseases. Trends Pharmacol Sci. 2021;42(4):255-267. doi:10.1016/j.tips.2021.01.003
Weiterführende Links:
Seltene Krankheiten in Europa: www.eurordis.org
Tag der Seltenen Krankheiten: https://www.rarediseaseday.org/
Verein NF Kinder: www.nfkinder.at



Hallo Katrin!
Super geschriebener Artikel, der die Problematik von Neurofibromatose beeindruckend aufzeigt. Umso mehr ist es wichtig, Forschungsgelder für diese Art von Krankheit zur Verfügung zu stellen. Dem kleinen Kenan und allen Betroffenen wünsche ich, dass der Forschung in absehbarer Zeit ein Durchbruch auf diesem Gebiet gelingt.
Mama
Hallo Katrin,
Gratuliere zu diesem hervorragenden Artikel. Leider gibt es eine ganze Reihe von Erkrankungen, die noch nicht heilbar sind, bzw. deren Entstehung ein Rätsel ist. Du kennst ja meine Präkollapse, die weder neurologische, noch kardiologisch erklärbar ist. Keine Therapie führt zu einer gravierenden Besserung.
Gleiches gilt - trotz intensiver Forschung - für bestimmte neurofibromatöse Erkrankungen.
Man kann also zur Zeit nur hoffen und versuchen, das Beste daraus zu machen.
Liebe Grüße
Dein Onkel Thomas